

Most people with HIV infection initially develop HIV-specific immune responses comprising HIV-specific CD8+ and CD4+ T cells and HIV-specific B cells and antibodies. These responses are suboptimal and ultimately fail in the majority of people, primarily because of viral immune evasion mechanisms. The preservation of strong HIV-specific antibody and cellular immune responses in people with non-progressive HIV infection does come at the expense of immune activation (see below - Long-term non-progression and elite controllers). This suggests that strategies to restore and maintain host immune responses will be important in the long-term management of patients with HIV disease and for cure strategies. Effective HIV-specific immune responses will also be necessary for the success of prophylactic and therapeutic HIV vaccines.
HIV-specific CD8+ T cell responses
CD8+ T cells recognise a variety of peptides from all HIV proteins including reverse transcriptase, envelope, core (gag) and the accessory proteins Vif and Nef, although CD8+ T responses are predominantly directed against epitopes encoded by the gag and env genes. CD8+ T cells recognise divergent HIV strains with greater frequency than HIV-specific CD4+ T cells. (Hammond et al., 1992) The control of HIV can be shown to be associated with CD8+ T cell responses (Northfield et al., 2007). Evidence for a direct role for CD8+ T cell responses in the control of HIV and simian immunodeficiency virus (SIV) infection is derived from animal models of macaques infected with SIV where depletion of CD8+ T cells results in rapid rebound of SIV viraemia. (Jin et al., 1999). Conversely, the induction of effector CD8+ memory T cells can control SIV infection (Hansen et al., 2016).
Measurement of CD8+ T cell responses
CD8+ T cell responses against HIV can be measured by a variety of assays (See section on Immunological diagnostics). Traditional chromium-release assays were used to detect antigen-specific CD8+ T cell killing of labelled targets. These assays require cell culture and are labour intensive. Other assays to detect antigen-specific T cells use flow cytometry and granule release assays, tetramers of MHC peptide, or intracellular cytokine production. Cytokine production can also be detected via enzyme-linked immunospot assay (ELISPOT). Potential discrepancies between the numbers of antigen-specific CD8+ T cells detected by tetramer staining and those detected by functional assays, such as chromium-release or intracellular cytokine staining, should be appreciated. Chromium-release assays require multiple ex vivo cell divisions and then detection by functional analysis. This may underestimate actual numbers of antigen-specific CD8+ T cells as these cells may readily apoptose ex vivo. Tetramer assays, on the other hand, may over-estimate CD8+ T cell responses by detecting cells with a TcR binding a specific antigen-MHC complex, which exhibit low cytokine production or cytolytic activity. Other methods for measuring CD8+ T cell-mediated killing include CD107a degranulation, fluorescence-based target elimination and caspase cleavage assays, each with advantages and disadvantages (Makedonas and Betts, 2011).
Role of CD8+ T cells in the pathogenesis of HIV disease
CD8+ T cells play an important role in delaying HIV disease progression. HIV disease progression is associated with a decline in HIV-specific CD8+ T-cell activity in some patients and slow progression of HIV disease is associated with strong CD8+ T-cell activity, upregulation of perforin expression and with polyfunctionality (Makedonas and Betts, 2011).
Furthermore, an inverse correlation between CD8+ T responses and plasma viral loads suggests a protective role for CD8+ T cells. (Chouquet et al., 2002) More direct evidence of a protective role of CD8+ T-cell responses in HIV infection comes from animal models. Monkeys subjected to CD8+ T-cell depletion cannot control plasma viraemia during primary SIV infection. (Schmitz et al., 1999) Furthermore, CD8+ T cell depletion during chronic SIV infection leads to profound increases in plasma viraemia. (Jin et al., 1999) and adoptive transfer of CD8= T cells at the time of infection reduces the viral reservoir (Ayala et al., 2016).
Human leukocyte antigen associations
The association between HLA genotype and HIV disease progression underscores the importance of CD8+ T-cell responses in HIV disease pathogenesis.(Goulder and Walker, 2012) HIV-specific CD8+ T cells recognise viral peptides when they are presented on the surface of virally-infected cells in association with HLA- I molecules. Different HLA molecules present different peptides. This variation thus influences the quality of the immune response. For example, HLA-B27 and HLA-B57 are associated with slow HIV disease progression. (Kaslow et al., 1996) Even a single amino acid change within HLA-B35 is associated with differences in HIV disease progression. (Gao et al., 2001) Studies on the evolution of viral reverse transcriptase and HLA types have demonstrated that HIV will adapt to HLA type at a population level. (Moore et al., 2002; Kawashima et al., 2009) Homozygosity of MHC-I molecule genes produces a more restricted CD8+ T cell response than heterozygosity, and has been associated with faster HIV disease progression. (Carrington et al., 1999)
An increase in HIV viral load has been associated with mutations in immunodominant epitopes of peptides eliciting CD8+ T-cell responses, commonly referred to as immune escape. Viral control depends on both the HLA, the nature of the TcR and function of the T cell. (Chen et al., 2012) In an individual, adapted and non-adapted epitopes differ in their recognition by CD8+ T cells but the presence of CD8+ T cells recognizing adapted epitopes can facilitate DC mediated transfer of virus between CD4+ T cells (Qin et al., 2019). This maintains persistence of HIV infected cells even in the presence of CTL responses.
The mechanism by which HLA molecules influence disease outcome differs at different stages of HIV disease. (Gao et al., 2005) For example, HLA-B57 is associated with an early effect in reducing CD4+ T-cell decline and has a persistent effect throughout the disease course. HLA-B27 has no benefit in the early decline in CD4+ T cell counts to < 200/μL but is important later in delaying disease progression. People with HLA-B35 have an accelerated loss of CD4+ T cells early in the disease course but CD4+ T-cell loss is not different to that seen in people with other HLA genotypes at later stages of disease. At least some of these effects are related to the generation of CTL, early suppression of viral replication and the fitness of escape mutants. (Ganusov et al., 2011) Although a small number of specific HLA alleles were initially described as impacting HIV viral load and disease outcome, most alleles can be shown to influence HIV viral load in an additive way. (Leslie et al., 2004)
HLA and KIR
While HLA-I molecules are important in the CD8+ T cell response, NK cells also contribute to viral clearance by responding to the absence or reduced expression of specific HLA-I molecules on virally infected cells. To recognize aberrant virus-infected cells, NK cells express both inhibitory and stimulatory molecules known as Killer Immunoglobulin Receptors or KIR (these molecules belong to the immunoglobulin superfamily of molecules). Polymorphic genes encode a range of KIR (Figure 8) and different haplotypes of KIR genes have been associated with differences in control of HIV replication and HIV disease outcome. (Martin and Carrington, 2013) The haplotypes containing 2DS activating receptors have been associated with elite control of HIV. (Malnati et al., 2017) The interaction between KIR and HLA-C is important as it represents the most frequent HLA molecule recognized by KIR and its expression is correlated with HIV control in different populations in large genome wide association studies. (Apps et al., 2013) The importance of HLA-C in control of HIV infection is evident in the HIV vpu downregulation of HLA-C in latently infected cells. (Bachtel et al., 2018) Such down regulation can be recognized by activating KIR. Successful combinations of HLA-KIR have been shown to control HIV in African populations. (Mori et al., 2019)
Figure 8. HLA-I and Killer Immunoglobulin Receptors (KIR)
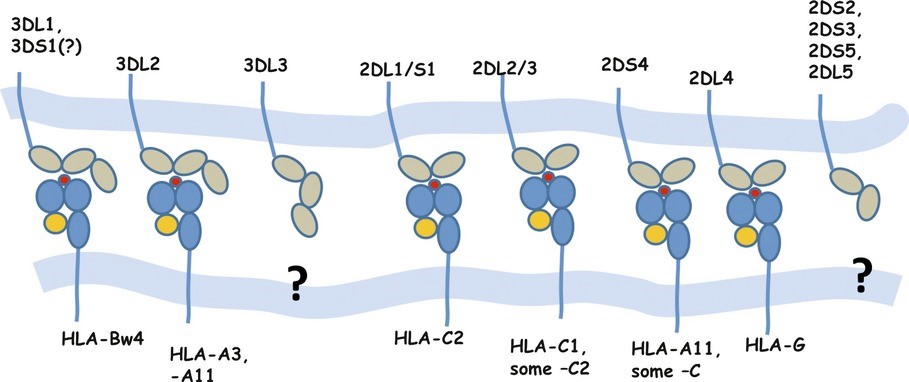
Note: Specific interactions between different KIR molecules on CD8+ T cells and NK cells and MHC-I molecules on target cells. HLA-C2 = HLA-C allotypes with Asn77/Lys80. HLA-C1 = HLA-C allotypes with Ser77/Asn80. Bw4 = HLA allotypes with the serologically defined Bw4 motif.
Source: Martin MP, Carrington M. Immunogenetics of HIV disease. Immunological Reviews. 2013; 254:245–64. Used with permission
Temporal change in CD8+ T-cell responses during HIV progression.
In the majority of people with HIV infection, HIV-specific CD8+ T cell responses are generated soon after the peak of viral replication during primary HIV infection. Indeed, the initial decrease in HIV viral load during primary infection is attributed to the generation of CD8+ T-cell responses. (Koup et al., 1994; Wilson et al., 2000) Detection of HIV-specific CD8+ T cell responses precedes the detection of HIV-specific neutralising antibodies.
Up to 10% of CD8+ T cells present during primary HIV infection are HIV-specific. (Wilson et al., 2000) In infections by other viruses, such as cytomegalovirus (CMV) and Epstein Barr virus (EBV), the proportion of virus-specific CD8+ T cells during chronic infection can be greater than 10%. The breadth of HIV-specific CD8+ T-cell responses in acute infection influences disease progression. Monoclonal expansion of HIV-specific CD8+ T cells in patients with acute HIV infection is associated with a poor prognosis.(Pantaleo et al., 1994) and a broad response favours control. (Radebe et al., 2014) Moreover, once the viral set point is reached following primary infection, there is an inverse relationship between the breadth of HIV-specific CD8+ T-cell responses and HIV viral load. (Chouquet et al., 2002)
HIV-specific CD8+ T-cell responses are detected in chronic phases of HIV disease in most patients with a frequency of up to 2% of total CD8+ T cells in blood (Ogg et al., 1998) and lymph nodes. (Connick et al., 1996) Antigen-specific CD8+ T cells have two fates. Some cells become terminally differentiated effector cells. Effector cells occur in large numbers but are not long-lived and die by apoptosis. In contrast, other CD8+ T cells become long-term memory cells that are in low numbers but are likely to persist. The CD8+ T-cell effector pool is continuously replenished from the CD8+ long-term memory T-cell pool. In turn, the CD8+ long-term memory T-cell pool is replenished from continued antigen stimulation of the naïve CD8+ T-cell pool. The expansion of CD8+ T cells is in large part dependent on continued antigen stimulation. This may explain why HIV-specific CD8+ T cells decline in parallel with HIV viral load reduction following combination antiretroviral therapy (cART).(Kalams et al., 1999b) When cART is implemented in the earliest stages of HIV infection, there is a very low frequency of infected cells and aborted production of CD8+ T cells (Ananworanich et al., 2014) and subsequent failure of control of HIV recrudescence after cART interuption. (Colby et al., 2018)
These observations highlight the problem of effective antiviral immune responses: a dependence on prolonged viral antigen exposure, which increases the viral reservoir. The failure of CTL clearance of residual latent virus infection in patients on suppressive cART depends on both the accumulation of viral escape mutants and the reduced HIV viral load attenuating the CTL responses. (Deng et al., 2015) Treatment interruption, however, is not effective in increasing CTL or viral control.
HIV-specific CD8+ T-cell functional abnormality
HIV-specific CD8+ T cells demonstrate a number of functional defects. (Migueles and Connors, 2015) CD8+ T cells are deficient in perforin, which is associated with poor ex vivo killing of targets when compared with CMV-specific CD8+ T cells in the same people. (Appay et al., 2000) Low levels of perforin have also been observed in CD8+ T cells from lymph nodes of people with HIV infection. (Andersson et al., 1999) Studies of elite controllers and long term non-progressors have provided insights into the factors that control effective immune responses to HIV; perforin production and polyfunctionality, in terms of broader range of cytokine production, being particularly important. Differences in maturation and expression of CD27 and CD28 may also be important and it has been found that most HIV-specific CD8+ T cells are CCR7-CD45RA- (pre-terminally differentiated) compared with CMV-specific CD8+ T cells which are CCR7-CD45RA+ (terminally differentiated). (Champagne et al., 2001) Failure of CD8+ T cells to mature could be the consequence of impaired CD4+ T-cell help. Disease progression after immune escape in HLA-B*5701-restricted epitopes is associated with the development of reduced perforin production and loss of polyfunctional T cells. (Buggert et al., 2014) Furthermore, HIV proteins specifically impair CD8+ T cell responses. Firstly, Nef and Tat decrease HLA class I expression on infected cells. Secondly, Nef increases Fas ligand (FasL) expression on virally infected cells leading to FasL- Fas interaction leading to apoptosis of CD8+ T cells. Finally, Nef inhibits apoptosis in the infected cell by blocking intracellular signalling events downstream of Fas. (Rajapaksa et al., 2012)
Non-cytolytic CD8+ T-cell responses
CD8+ T cells from people with HIV infection can inhibit the replication of HIV in tissue culture by a non-cytolytic mechanism. (Mackewicz and Levy, 1992) This activity has been attributed to the secretion of a cell-associated factor (CAF). CAF inhibits HIV replication by non-lytic mechanisms and does not impair the function of the infected cell; it does not alter the activation status of the CD4+ T cell or cause CD4+ T cell proliferation; it acts independently of MHC restriction; it is active against many strains of HIV-1, HIV-2 and SIV. CAF inhibits viral transcription by binding to HIV long-terminal repeat (LTR) of viral DNA (Copeland et al., 1995; Shridhar et al., 2014) interrupting the ability of HIV Tat to accelerate HIV transcription. The CAF inhibition of the transcription complex to reduce HIV expression is the core characteristic. (Blazek et al., 2016)
CAF activity correlates with clinical disease progression. High levels are seen in early HIV disease or non-progression. Patients with advanced HIV disease have low levels of CAF and a limited number of longitudinal studies have demonstrated that the loss of CAF is associated with disease progression, which resolves in people on cART.
While alpha-defensin and beta-chemokines have some CAF characteristics (Zhang et al., 2002), the exact nature of CAF remains unknown. Alpha-defensins and beta-chemokines are produced by cells other than CD8+ T cells, which was not a characteristic of the original description of CAF. A number of putative genes have been shown to be differentially expressed in CD8+ T cells showing high CAF activity (Martinez-Mariño et al., 2007).
HIV-specific CD4+ T-cell responses
HIV-specific CD4+ T-cell responses are generated during acute HIV infection in the vast majority of people with HIV infection but are markedly reduced or lost in the chronic stages of HIV disease. Suboptimal CD4+ T-cell help is central to the immunodeficiency of HIV disease. Preferential infection of HIV-specific cells may offer one explanation for poor HIV-specific CD4+ T cell responses. (Douek et al., 2002) Similar infection of antigen-specific T cells by HIV has been observed in people who received an HIV vaccine including the Ad5 adjuvant. (Hu et al., 2014) The depletion of CD4+ T-cells responding to HIV means that any immunological interventions to expand HIV-specific CD4+ T-cells will need to block HIV infection with cART.
Measurement of CD4+ T-cell responses
A variety of in vitro assays are available to measure CD4+ T-cell responses. Traditionally, CD4+ T-cell responses were measured using lymphocyte proliferation assays. In these assays, CD4+ T cells are mixed with HIV antigen and 3H-thymidine. CD4+ T cells which recognise HIV antigens proliferate and incorporate 3H-thymidine. The 3H-thymidine incorporation gives a qualitative measure of antigen-specific CD4+ T cell responses.
Alternatively, CD4+ T-cell responses can be measured by the detection of cytokine production following stimulation with HIV antigens. This may be measured either by ELISPOT assay or by intracellular staining and flow cytometry. These assays give qualitative and quantitative results. In contrast to CD8+ T-cell responses, measurement of CD4+ T-cell responses by tetramer assays has limited availability because of lower affinity of peptide binding to MHC-II molecules compared to MHC-I molecules. Another method for detection and isolation of antigen specific CD4+ T cells uses the expression of OX40 (CD134) and CD25 shortly after antigen stimulation as a marker of antigen-activated cells. (Zaunders et al., 2009)
Role of CD4+ T-cell depletion and dysfunction in HIV pathogenesis
Loss of effective CD4+ T-cell responses has detrimental consequences for both CD8+ T cell and B-cell responses against HIV. CD4+ T-cell help is crucial for priming CD8+ T cells, (Ridge et al., 1998) maintaining CD8+ T cell memory (Del Cornò et al., 2005), and maturing CD8+ T cell function (Figure 9). CD4+ T cells provide this help through the production of local cytokines, such as IL-2 and IL-15, which stimulate CD8+ T-cell activity. CD4+ T cells also enhance co-stimulatory pathways between antigen-presenting cells and cytolytic CD8+ T cells via upregulation of CD40L expression. CD40L is expressed on activated CD4+ T cells. CD40L is crucial in triggering DC to produce IL-12 which, in turn, is central to the initiation of CD8+ T-cell responses. Some viruses can by-pass this step by activation of the DC directly but HIV reduces innate signalling, including interferon signalling in myeloid DC. Activation of CD8+ T cells by DC then depends on providing DC-T cell signalling by plasmacytoid DCs that can be directly activated by HIV and produce high levels of IFNα and other type I interferons (Del Cornò et al., 2005), which can then activate the myeloid DC and initiate CD8+ T cell responses. HIV can interupt this process both by reducing plasmacytoid DC numbers and function and by blocking innate signalling pathways in myeloid DC.
Figure 9. The relationship between CD4+ helper T-cell function and CD8+ T-cell activity
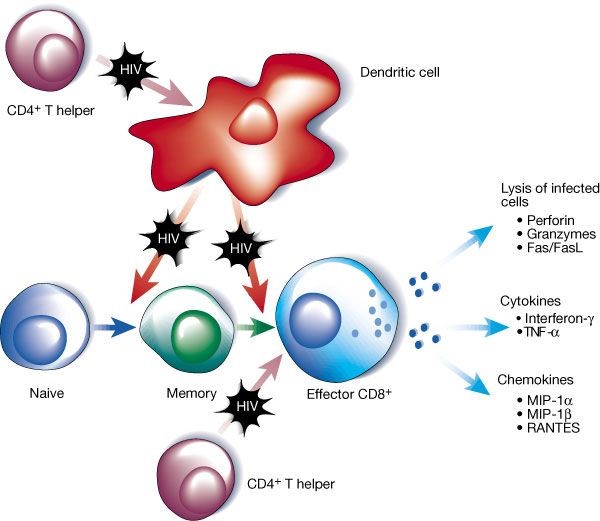
Note: CD4+ T cells are important for priming dendritic cells to initiate CD8+ T cell responses. They help maintain memory T cells and are important in maturation of CD8+ T-cell function. All of these actions are impaired by HIV infection. In addition, HIV can directly impair dendritic cell function. FasL= Fas ligand; MIP= macrophage inflammatory proteins; RANTES= regulated on activation normal T cell expressed and secreted; TNF= tumour necrosis factor.
Source: McMichael AJ, Rowland-Jones SL. Cellular Immune responses to HIV. Nature 2001; 410:980-7. Used with permission.
Temporal change in CD4+ T-cell responses during HIV progression
HIV-specific CD4+ T-cell responses are generated early following infection with HIV and are detectable in people treated early in acute infection. (Rosenberg et al., 1997) In contrast, HIV-specific CD4+ T-cell responses are low or absent in most people with chronic HIV disease, including those on effective cART, but are detectable in people with long-term non-progressive disease. (Saez-Cirion et al., 2014) While HIV-specific CD4+ T-cell responses have been detected in most people with established HIV disease, they have fewer HIV-specific than CMV-specific CD4+ T cells, suggesting a limited HIV-specific CD4+ T-cell response. (Pitcher et al., 1999) The decline in HIV-specific CD4+ T-cell responses is a fundamental process in HIV disease progression.
Several factors are thought to contribute to the impairment of HIV-specific CD4+ T-cell responses in progressive HIV disease. These include virus-induced anergy, antigen-induced cell death, direct infection and pyroptosis or apoptosis of CD4+ T cells. HIV Tat down regulates HLA-II expression and therefore may lead to viral induced anergy by impairing antigen recognition.(Douek et al., 2002; Kanazawa et al., 2000; Paroli et al., 2001; Kalams et al., 1999a) An inverse correlation between HIV-specific CD4+ T-cell responses (Rosenberg et al., 1997; Kalams and Walker, 1998) and plasma HIV viral load suggests that HIV-specific CD4+ T-cell responses are protective. However, a causal relationship has not been proven. HIV-specific CD4+ T-cell responses decrease following control of HIV replication with cART, possibly because of reduced antigen presentation. (Autran et al., 1997) HIV-specific CD4+ T-cell responses are preserved, however, in people who commence cART at low HIV viral loads. (Oxenius et al., 2002) There is also abnormal production of T-cell subsets from naïve T cells during HIV infection. (DaFonseca et al., 2015)
CD4+ T cell defects in HIV disease
Progressive HIV disease is characterised by quantitative and qualitative defects in CD4+ T cells manifesting as a decline in both CD4+ T-cell numbers and function. The proximal cause of CD4+ T cell loss in HIV disease is a perturbation of CD4+ T-cell homeostasis. This perturbation results from both an increase in the destruction of mature effector memory CD4+ T cells and decreased replacement of these cells with precursor cells, including central memory CD4+ T cells and immature progenitors. Other factors contributing to the loss of CD4+ T cells in people with HIV infection include generalised immune activation leading to enhanced T-cell proliferation and death, direct cytopathic effects of HIV and altered trafficking with redistribution of CD4+ T cells to lymphoid organs, including the gut lymphoid tissue. A number of factors contribute to the impaired function of CD4+ T cells in people with HIV disease. These include reductions in the proportion of naïve CD4+ T cells, restriction of T-cell receptor repertoire and HIV-induced anergy.
Quantitative defects in CD4+ T cells
CD4+ T-cell homeostasis refers to the process of balance between cell production and destruction resulting in maintenance of stable CD4+ T-cell numbers. Processes which result in new CD4+ T-cell production are referred to as entry pathways and processes which result in the destruction of CD4+ T cells are referred to as exit pathways. These processes are usually tightly regulated, but perturbation of this process leads to CD4+ T cell decreases in people with HIV disease (Figure 2). Predominant entry pathways include CD4+ T-cell production by proliferation of peripheral CD4+ T cells or by differentiation from progenitors via thymic (or extrathymic) pathways. The predominant exit pathways include enhanced proliferation and cell death and HIV-mediated CD4+ T-cell destruction. Over time in untreated people, exit pathways predominate, resulting in a median CD4+ T-cell decline at a rate of 84 cells/μL per year. (Lang et al., 1989)
Alterations to entry pathways
Increased CD4+ T cell proliferation
Untreated HIV infection is characterised by marked increases in immune activation and lymphocyte proliferation, which exceed that observed in people without HIV infection by at least two- to three-fold. (Vrisekoop et al., 2015) During HIV infection, naive CD4+ T cells live about 618 days and CD8+ T cells about 271days. Memory CD4+ T cells live about 53 days and CD8+ T cells about 43 days. This is about 3-fold shorter than healthy controls and after commencing cART the lifespan remains half that of controls. The augmented proliferation is predominantly observed in memory CD4+ (and CD8+) T cells, which proliferate rapidly in comparison to naïve T cells.(Kovacs et al., 2001) Although gut derived microbial products may be a predominant driver of proliferation (see Immune activation below) other causes of peripheral lymphocyte proliferation include: HIV-antigen specific expansion; stimulation of T cells by cross-reactive antigens; and cytokine mediated bystander activation (Hazenberg et al., 2000). Activation-induced apoptosis leads to eventual decline in CD4+ T-cell numbers, if not compensated for by other entry pathways (see below). Increased lymphocyte proliferation induces anergy (see below).
Reduced CD4+ T cell differentiation from progenitor cells
Contrary to previously held beliefs that the thymus is not active in adults, many reports demonstrate that substantial thymic output persists even into late adulthood. (Douek et al., 1998) However, the role of the thymus in adults with HIV infection is poorly understood. Thymic function may be measured by imaging (e.g. CT scan) or measurement of T-cell receptor excision circles (TREC, i.e. episomal circular DNA products produced following T-cell receptor gene rearrangement during thymic development) in blood T cells. When peripheral CD4+ T cell proliferation is taken into account, these measures of thymic function suggest that thymic function is reduced in people with HIV disease. While thymic size is decreased in people with advanced HIV disease, people with moderate HIV-related immune deficiency have more thymic tissue than age-matched controls without HIV infection.(McCune et al., 1998) Furthermore, the amount of thymic tissue correlates with the rate of increase in naïve CD4+ T cells following initiation of cART. (Smith et al., 2000) These findings suggest that increased production of naïve CD4+ T cells by the thymus in people with early HIV disease initially compensates for the loss of CD4+ T cells secondary to increased peripheral turnover. However, thymic function wanes over time, perhaps because of viral factors discussed below, which leads to a net decrease in the size of the CD4+ T-cell pool.
Alterations to exit pathways
HIV-induced CD4+ T-cell loss in blood and tissues
HIV induces cell death in both infected and uninfected bystander CD4+ T cells by cytopathic, apoptotic and immune processes. Mechanisms underlying HIV-induced CD4+ T-cell loss are outlined in Table 1.
Despite a marked increase in CD4+ T-cell turnover in HIV disease, direct HIV cytopathic effect on CD4+ T cells in blood has been difficult to demonstrate. Studies of gastrointestinal tract lymphoid cells, however, have shown a rapid and profound loss of CD4+ T cells in acute HIV infection in humans (Mehandru et al., 2004) and SIV infection in primates. (Veazey et al., 2005; Mattapallil et al., 2005) Memory CD4+ T cells are lost primarily in the lamina propria of the gut mucosa. (Brenchley et al., 2004; Li et al., 2005) This happens as early as days 4 to 14 of acute infection. Less than 20% of the cells are lost by direct infection or by CTL activity. The predominant mechanism of cell loss is a bystander effect by gp120 induced FasL-mediated apoptosis and is specific for CD4+ T cells. CD4+ T-cell depletion in the gut has been demonstrated at all stages of HIV disease and accounts for a significant and sustained loss of total body CD4+ T cells. (Brenchley et al., 2004) The gut depletion does not occur in long term non-progressors who maintain CD4+ T cells and suppress HIV replication (Ciccone et al., 2011)
Much of the CD4+ T cell loss seen in HIV infection has been attributed to pyroptosis (Doitsh et al., 2014), a process of cell death mediated by caspase 1 and associated with production of IL-1b and IL-18. Peripheral blood CD4+ T cells are not susceptible to pyroptosis (Muñoz-Arias et al., 2015) suggesting that this pathway is most active in tissue CD4+ T cells. Most of this loss is attributed to abortive infection and triggering of innate pathways by incomplete DNA transcripts. (Monroe et al., 2014) The effect of cART on depletion of CD4+ memory T cells in the gastrointestinal tract is variable, although reconstitution is more effective following treatment in primary infection,(George et al., 2005; Schuetz et al., 2014) and reconstitution of gut Th17 cells that maintain mucosal integrity may also occur with therapy. (d’Ettorre et al., 2014)
Table 1. Mechanism of CD4 cell destruction in HIV disease
|
Cell |
Process |
Mechanism |
|
Infected |
Cytopathology |
Disruption of cell membrane syncytia formation Accumulation of unintegrated viral DNA |
|
Apoptosis |
Env and Vpr-induced apoptosis, Tat-induced Fas expression |
|
|
Immunological |
HIV-specific CTL |
|
|
NK cell killing |
||
|
Bystander |
Cytopathology |
Syncytia formation |
|
Apoptosis |
Tat and Nef increase FasL expression |
|
|
Immunological |
Autoimmune processes, ADCC |
ADCC = antibody-dependent cellular cytotoxicity; CTL = cytotoxic T lymphocyte.
Adapted from McCune JM. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 2001; 410:974-79.
Redistribution of CD4+ T cells
Alterations to physiological pathways of lymphocyte trafficking may account for some changes observed in the peripheral blood of people with HIV infection. CD4+ T cells are sequestered in lymphoid tissues, including lymph nodes, of people with untreated HIV infection. Antigenic stimuli within lymph nodes also result in the activation and retention of CD4+ T cells within the lymphoid tissues. Increased inflammation, including chemokine expression, and fibrosis in the lymph nodes of people with untreated HIV disease may contribute to this phenomenon.(Zeng et al., 2011; Tedla et al., 1996) The initial phase of CD4+ T cell increase following initiation of cART is thought to represent the corresponding redistribution of CD4+ T cells from lymphoid tissues into the peripheral blood. (Pakker et al., 1998)
Qualitative defects in CD4+ T cell function in HIV disease
Memory and naïve T cells
In addition to the quantitative defects, there are many qualitative defects in CD4+ T cells in progressive HIV disease. There is a decrease in the proportion of naïve CD4+ T cells (CD4+CD45RA+CD62L+) and a corresponding increase in the proportion of memory CD4+ T cells (CD4+CD45RO+). This alteration may be secondary to reduced thymic production of naïve CD4+ T cells and an increased peripheral proliferation of memory CD4+ T cells. Preferential HIV infection of naïve CD4+ T cells with cytopathic HIV variants may also contribute to the relative decrease in naïve CD4+ T-cell numbers. There is also a restriction in the TcR repertoire (Gorochov et al., 1998) as a result of deletion of some antigen-specific CD4+ T-cell clones following antigen-induced cell death.
CD4+ T-cell function in people with HIV infection is also largely limited by anergy, functionally defined as a state of hyporesponsiveness in which lymphocytes do not respond appropriately following antigenic stimulation. Anergy is present at all stages of HIV disease and can be demonstrated before any decline in CD4+ T-cell counts. It can be measured in vivo by skin testing and in vitro by antigen stimulation assays. Anergy is a state of unresponsiveness that results from a number of integrated signals including those from suboptimal TcR stimulation, reduced co-stimulation, increased Treg signalling and negative regulators or immune check points, such as PD-1. (Day et al., 2006)
One other recently defined mechanism for induction of anergy is TLR7 signalling by chronic infection and recognition of viral products. (Dominguez-Villar et al., 2015) While the mechanisms underlying anergic processes in HIV infection are not completely defined, many are reduced following the initiation of cART. (Autran et al., 1997)
In HIV infection, there is altered expression of T-cell molecules that leads to an impaired antigen-specific response, commonly referred to as T-cell exhaustion. Increased expression of CTLA4 (Kaufmann et al., 2007) and PD-1 (Day et al., 2006) on both CD4+ and CD8+ T cells contribute to reduced immune responses in people with HIV infection. Blocking the PD-1 pathway allows recovery of CTL responses. (Wykes and Lewin, 2018)
T follicular helper cells
Recent work has suggested that Tfh cells are a CD4+ T cell population that is susceptible to HIV infection and may be a reservoir of persistent HIV infection during cART. (Perreau et al., 2013) In the lymph node, Tfh cells express the immune checkpoint inhibitor PD-1 and the chemokine receptor CXCR5. The presence of circulating precursors for these cells correlates with the production of broadly neutralizing antibodies to HIV (Locci et al., 2013). However, the B cell follicle is a site of persistent HIV infection. (Banga et al., 2016) Seeding of the peripheral blood from this reservoir may also occur since the blood viral reservoir of HIV is enriched in cells expressing the immune checkpoint PD-1 (Fromentin et al., 2019). Within the follicles, follicular regulatory T cells (Tfr) are susceptible to infection by CCR5-tropic HIV (Miller et al., 2017) and a population of T follicular regulatory CD8+ T cells inhibits the germinal centre reaction and HIV antibody production. (Miles et al., 2016)
Th17 cells
CD4+ T cells are depleted from the gut mucosa early in HIV infection. Most of these CD4+ T cells have the phenotype of Th17 cells. Depletion of other populations including antigen-presenting cells and ILC also occurs (Mudd and Brenchley, 2016) The gut homing T cells express the chemokine receptors CCR6 and CXCR3 and is the population of peripheral blood CD4+ T cells most frequently infected cells in individuals on suppressive cART. (Khoury et al., 2016) Microbial translocation is increased as a consequene of loss of mucosal integrity. Loss of IL-17 producing ILC17 occurs in SIV infection (Xu et al., 2012) Early cART can reduce the depletion of Th17 cells but in humans even initiation of cART in acute infection is associated with some depletion of Th17 cells. (Schuetz et al., 2014)
Regulatory T cells
Treg cells are also important in regulating antigen-specific immune responses. Treg cells are generated in the thymus or in the periphery and are marked by expression of the intracellular transcription factor FoxP3. Treg cell numbers are regulated in the thymus by DC, in particular those developing in the presence of thymic stromal lymphopoetin (TSLP). Treg cells express CD4, CD25 and intracellular FoxP3, have reduced surface expression of CD127, are susceptible to HIV infection and may be depleted from the gut early after infection. (Oswald-Richter et al., 2004; Chase et al., 2007) Loss of Treg cells may be associated with increased T-cell proliferation (Eggena et al., 2005), although this has not been a consistent finding. (Lim et al., 2007) The persistence or expansion of antigen specific Treg cells may block antigen specific CTL. (Legrand et al., 2006)
T stem cell memory
Stem memory T cells (Tscm cells) represent a small population of long-lived memory cells with high proliferative capacity. (Gattinoni et al., 2011) These cells express the marker CD45RA but represent a population with high frequency of HIV infection. (Buzon et al., 2014) The proportion of these cells in HIV infected individuals is a predictor of immune function (Lu et al., 2017)
Antibody responses against HIV
All HIV-infected people generate antibody responses against HIV. While the clinical relevance of these antibodies in established HIV infection remains to be defined, the generation of antibodies for prophylactic vaccine strategies is thought to be critical. The production of effective broadly neutralising monoclonal antibodies and the mechanisms for their generation is seen as the pathway to more effective antibodies. (Burton et al., 2012; Wang et al., 2015)
HIV-specific antibodies
Currently, HIV infection is diagnosed by detection of HIV antibodies using enzyme-linked immunosorbent assays (ELISAs). Currently used ELISAs allow identification of antibodies that bind to HIV antigens, in addition to detecting HIV antigens, specifically HIV p24. Antibodies to HIV are characterised further by demonstrating which viral proteins the antibodies bind to using Western blot assays. HIV antibodies detected by ELISA and confirmatory Western blot assay may not have functional activity, as opposed to neutralising antibodies that inactivate or neutralise HIV replication. The appearance of HIV-binding antibodies detected by the ELISA and Western blot assay (see section Virological diagnostics) occurs prior to the appearance of neutralising antibodies. The first HIV-specific antibodies detected are those directed against the structural, or gag, proteins of HIV, p24 and p17, and the gag precursor, p55. The development of antibodies to p24 is associated with a decrease in the serum levels of free p24 antigen. Antibodies to the gag proteins are
followed by the appearance of antibodies to the envelope proteins (gp160, gp120, p88 and gp41) and to the products of the pol gene (p31, p51 and p66).
Broadly neutralising antibodies
Broadly neutralising antibodies are those reactive with a broad range of virus isolates. These can be generated in many subjects, including some natural controllers of HIV infection, but they are generally related to high HIV viral load and to chronic antigen exposure and develop later in the course of infection. A number of these antibodies have been generated as monoclonal antibodies and used therapeutically to control or block infection in non-human primates (Hessell et al., 2009; Scheid et al., 2011) and humans (Caskey et al., 2015; Halper-Stromberg et al., 2014; Caskey et al., 2019) Therapeutically, it is evident that combinations of monoclonal antibodies are more effective than a single monoclonal antibody suggesting immune escape may be important when antibody concentration is declining (Caskey et al., 2019) Therapeutic antibodies include those that bind to the CD4 binding site, the membrane proximal region binding to gp41, glycan and amino acid residues in the V1, V2 and V3 regions of the HIV env.(Scheid et al., 2011)
These monoclonal antibodies require the ability to bind to Fc receptors on cells for optimal efficacy (Hessell et al., 2007; Bournazos et al., 2014) suggesting that they may be functional by mechanisms involving cells bearing receptors of the Fc regions of IgG antibodies (Fc-gamma receptors or FcgRs), such as antibody-dependent cellular cytotoxicity (ADCC). Furthermore, it is now clear that broadly neutralising antibodies have the ability to bind to the gp120 trimer compared to non-neutralising gp120 antibodies that bind to monomeric gp120. (Burton and Mascola, 2015) Recent work has focused on the pathways for the development of such broadly neutralising antibodies. These broadly reactive antibodies characteristically have long complementarity determining regions (CDRs) and are hypermutated, indicating that they depend on effective germinal centre reactions and the activity of Tfh cells. (Gao et al., 2014; Locci et al., 2013) Sequential vaccination strategies have been proposed that may lead to the development of such antibodies in vaccinated people. (Alter and Barouch, 2015)
HIV-specific neutralising antibodies bind to HIV and inactivate it
Several weeks after the decrease in HIV viraemia following primary infection, neutralising antibodies are detectable, and primarily target proteins expressed on the viral envelope. (Koup et al., 1994) There are conflicting data concerning the correlation of HIV-specific neutralising antibodies and HIV disease progression. (Yang and Walker, 1997; Lifson et al., 1991; Pantaleo et al., 1995) Some studies have demonstrated that people with long-term non-progressive HIV disease have high levels of antibodies that are capable of neutralising a large number of HIV isolates. It has been suggested that constantly changing viral quasi-species in this patient group may give rise to a wide range of epitopes for antigen presentation. (Delwart et al., 1994) In contrast, people with progressive HIV disease have low or absent antibodies, which neutralise only autologous HIV strains. (Lathey et al., 1999) In addition, the development of neutralising antibodies was associated with a reduction in HIV viral load in health-care workers who acquired HIV infection following needlestick injuries. (Lathey et al., 1999) and passive administration of neutralising antibodies protected macaques from SIV infection (Parren et al., 2001) and reduced viral rebound after cessation of cART in humans with HIV infection. (Caskey et al., 2019) There is disagreement in the literature regarding any protective effect of maternal neutralising antibodies in HIV-exposed infants.(Lathey et al., 1999; Mabondzo et al., 1998) Also, neutralising antibodies have not been detected in people who are persistently exposed to HIV but remain uninfected (highly exposed seronegative [HESN] individuals.(Hogan and Hammer, 2001)
A number of factors limit the development of effective HIV-specific neutralising antibodies. These include the poor antigenicity of HIV-envelope proteins and the fact that critical sites, such as the CD4 and co-receptor binding site, are hidden as a result of glycosylation of the complex three dimensional structure of gp120. Broadly neutralising antibodies inhibit viral function in three ways: they induce the dissociation of gp120 from gp41; they directly inhibit viral binding to receptor- coreceptor complexes; and they interfere with post-attachment (Parren et al., 2001) steps of gp41, which lead to virus-membrane fusion. Human studies have confirmed the importance of the membrane proximal region of env as a target resistant to viral escape. (Trkola et al., 2005)
Antibody-dependent cellular cytotoxicity (ADCC)
Antibodies to both gp120 and gp41 participate in antibody-dependent cellular cytotoxicity (ADCC)-mediated killing of HIV-infected cells.(Evans et al., 1989) The concentration of anti-envelope antibodies capable of mediating ADCC is highest in the early stages of HIV infection. ADCC-mediated cytotoxicity correlates with control of HIV viraemia during acute infection. (Connick et al., 1996) The protective effect of neutralising antibodies in acute infection depends on binding to FcgR but not complement activation. Thus, IgG antibodies to HIV envelope antigens exert a protective effect by both clearance of cell-free virus and clearance of HIV-infected cells binding antibody by mechanisms involving ADCC.(Hessell et al., 2007)
Other cells and contributors to HIV pathogenesis
Lymph node structure
There are three clearly defined stages of lymph node destruction in HIV disease: follicular hyperplasia; follicular disruption; and follicular depletion. High levels of HIV RNA are present in the lymph nodes at all stages of HIV disease. (Embretson et al., 1993)
In the first stage, follicular hyperplasia (type I), the lymph node has the appearance of a normally reactive node. The lymphoid follicles enlarge and coalesce and are associated with markedly hyperplastic germinal centres. HIV RNA is confined within the germinal centres. HIV is attached to follicular DC via antibody in immune complexes associated with complement. HIV-induced follicular expansion is characterised by the predominant expansion of B cells associated with Tfh cells, which are a target for HIV infection and may be a reservoir for HIV replication.(Perreau et al., 2013) Follicular hyperplasia is generally associated with blood CD4+ T cell counts > 500/μL.
The next stage, follicular disruption (type II), is characterised by gradual loss of germinal centres, thickening of the lymph node capsule and irregularly distributed proliferation of small blood vessels. Nodal architecture is distorted with a polymorphic population of lymphoid cells, plasma cells and multinucleated giant cells. HIV replication is no longer contained within the germinal centres. Follicular disruption generally correlates with a CD4+ T cell count of 200-500/μL.
In the third stage, follicular disruption (type III), the germinal centres are completely involuted and the lymph node architecture completely destroyed. There is striking lymphocytic depletion and reduction in lymph node size. Excessive vascularisation, diffuse fibrosis and plasma-cell infiltration characterise the inter-follicular regions. Appropriate T-cell responses are decreased in late-stage HIV disease secondary to the destruction of the stromal microenvironment of lymph nodes.(Estes, 2013)
The use of inhibitors of fibrosis have been shown to be effective in reducing T-cell loss in non-human primates (Estes et al., 2015) but have not shown efficacy in human clinical trials.
Monocyte-derived macrophages
HIV infection of monocyte-derived macrophages leads to sustained infection with limited cytopathic effect.(Collman et al., 1989) Monocyte/macrophage infection contributes only 1% to the overall viral load in untreated people and is rarely detected on ART.(Ho et al., 1995; Cattin et al., 2019) However, monocyte/macrophages play a critical role in the pathogenesis of HIV disease largely as a result of impaired function. HIV-infected macrophages are an important reservoir in people on cART. HIV-infected monocytes/macrophages as well as DC contribute to CD4+ T-cell depletion by multiple means, including: gp-120 interactions with CD4 molecules on neighbouring T cells resulting in cellular fusion, increased FasL expression; and HIV infection of neighbouring CD4+ T cells. HIV infection of macrophages and microglia are important in the pathogenesis of HIV encephalopathy. Furthermore, HIV impairs macrophage-effector functions, thereby contributing to development of opportunistic infections, such as toxoplasmosis and candidiasis. Monocytes/macrophages from people with HIV infection have reduced expression of MHC-II and co-stimulatory molecules, which impair antigen presentation, and downregulation of macrophage-surface receptors (e.g. mannose receptor) likely impairs internalisation of pathogens. (Caldwell et al., 2000)
Monocyte subpopulations that express low CD14 and high CD16 have the ability to produce higher levels of pro-inflammatory cytokines. This population of cells is more susceptible to HIV infection than CD14 high monocytes in vitro and is more frequently infected during HIV infection in vivo. (Ellery et al., 2007) These cells may be the predominant monocyte population that is recruited into the central nervous system (CNS) and contribute to the development of the HIV-associated neurological disease.
Natural Killer cells and gamma-delta T cells.
NK cells are activated in the absence of MHC-I and provide protection against virus infections when CTL responses are ineffective. Loss of MHC-- molecules, such as occurs in HIV infected cells producing Nef, will result in NK targeting and cytolysis by mechanisms shared with CD8+ T cells. (Brown et al., 2005) HLA-E is expressed specifically on infected CD4+ T cells and is recognised by gamma-delta T cells. (Fausther-Bovendo et al., 2008)
NK cell activation is associated with expression of TRAIL, a TNF family molecule that induces apoptotic cell death in TRAIL-ligand expressing target cells. NK cells also have an immunoregulatory effect as they are responsible for the final destruction of DC in lymph nodes and thus will limit the expansion of T cells.
B cells
HIV infection is associated with abnormal activation of B cells.(Jacobson et al., 1991) Multiple viral proteins, in particular gp41, induce polyclonal B cell activation.(Chirmule et al., 1990) Hypergammaglobulinemia develops and is particularly evident in children.(Ammann and Levy, 1986) Polyclonal B cell hyperactivity during the early phases of HIV infection may be the major process predisposing to B cell malignant transformation and AIDS-associated lymphomas.
Summary
HIV-specific CD8+ T cells provide the cornerstone of the host immune response against HIV. HIV-specific CD8+ T-cell responses are generated early in acute infection and persist into the chronic phases of HIV disease. HIV-specific CD8+ T-cell responses are directed against a broad range of HIV peptides and have been demonstrated to control SIV viraemia in animal models. The influence of HLA-I on HIV disease progression underscores the importance of CD8+ T-cell responses in HIV disease pathogenesis. In contrast, HIV-specific CD4+ T-cell responses, while being generated in the acute stages of HIV disease, are absent or severely impaired in the vast majority of people with chronic HIV disease. Impairment of HIV-specific CD4+ T-cell responses is one factor that leads to the waning of HIV-specific CD8+ T cells. The contribution of HIV-specific antibodies to the control of HIV infection is less clear. While HIV-specific neutralising antibodies are detected in most people, these antibodies rarely neutralise concurrent autologous strains of HIV. Nonetheless, given the capacity of passive administration of broadly neutralising antibodies to protect against infection in animal models and in humans, the generation of HIV-specific neutralising antibodies is seen as a critical component of successful prophylactic vaccine strategies.